November 22, 2006
Axon Dynamics Workshop: Window into ALS Disease Process
[QUICK SUMMARY: Molecular events within the axon, the long fiber from motor neurons that die in ALS, served as the focus of a workshop sponsored by The ALS Association. Experts who can inform on the normal processes within the motor neuron met with ALS researchers in order to better grasp the basis of the disease at the cellular level and how to best target candidate therapies.]
The workshop, Axon Dynamics and Synaptic Junctions, funded by the Greater New York Chapter of The Association and held at Banbury Center, Cold Spring Harbor, NY, focused on the most recent progress in appreciating the extraordinarily complex output fiber of nerve cells called the axon. The axon is what enables nerve cells to convert the electrical charge of their activation into a chemical message released nearly instantly. For motor neurons, that conversion takes place as much as a meter away to contract muscle. This represents an extreme challenge for a single microscopic cell and is the very signal interrupted by the disease process in ALS.
 |
|
Copyright David G. King, 2006. Used with permission |
Researchers find in a mouse model of ALS that the earliest event heralding the disease is a retreat of the axon terminals from their junction at muscle. This was strikingly illustrated by new microscopic techniques on transgenic mice employed by Jeff Lichtman of Harvard, who had presented at the stem cell workshop last year at Banbury. Also at this year’s workshop, Swiss investigator Pico Caroni, Ph.D., at the Miescher Institute in Basel revealed that certain genes are changing their rate of making protein even before that first detachment of the axons takes place, well before symptoms appear in the mouse. His investigations have revealed a detailed time course for how axons change very early in the mutant mouse model of ALS. Once confirmed that such changes are relevant to the disease, the details could provide targets for therapeutic effort.
 |
| Photo Credit: Miriam Chua |
These are two examples of how researchers at the workshop provided the emerging ideas that will guide renewed efforts and future collaborations. Investigators revisited and expanded on how axonal traffic moves materials from the protein factories in the cell body out to the terminals. At the same time, the axon collects and conveys back from muscle junction many signal and helper molecules that recalibrate and sustain the cell.
Players within the axon include the motor proteins that move materials, the tracks along which the motors advance, as well as the packaging of the materials. The analogy offered by Chris Miller, Ph.D., of King’s College, London, is that of locomotives, cargo cars, and rails. ALS is the train wreck, but what caused the accident? It could be a stalled engine, bent rails or spent fuel.
Dynein and Dynactin form a Molecular Motor
Erika Holzbaur, Ph.D., at the University of Pennsylvania, a workshop co-organizer, detailed her investigations into components of motor molecules in the axon, which include proteins called dynein and dynactin. Changes to these proteins can give motor neuron symptoms similar to ALS. A dynactin mutation that produces human motor neuron disease, introduced into mice, was described by Philip Wong, Ph.D., of Johns Hopkins in Baltimore. Wong showed the neurons in this mouse apparently cannot cart off their trash. Microscopic packages of cell debris accumulate in these cells well before symptom onset in the mice.
Scaffolding and Tracks for Axonal Transport
Don Cleveland, Ph.D., of the University of California, San Diego, and a co-organizer of the workshop, spoke on the neurofilaments, scaffold proteins that link to form the inner structure of the axon. These neurofilament proteins create a supportive meshwork within the fiber. Surprisingly, removing part of the neurofilament linkers, and even removing neurofilaments altogether, extends survival by two months of the mutant SOD1 transgenic mouse. These mice express the human mutant protein, copper-zinc superoxide dismutase involved in some inherited forms of ALS.
Miller showed that many different mutant SOD1 molecules--more than 117 to date are known to cause ALS--can slow axonal transport. The result is congestion of the cell power suppliers, the mitochondria.
Mitochondria and Synaptic Protein: Key Cargoes for Axonal Motors
Mitochondria produce the energy that drives all cell processes such as putting together proteins and bringing them to their proper destinations. The mitochondria themselves must be able to get to the far endings of nerves. Key to delivery of mitochondria may be particular proteins that hook cargoes to the
motor molecules as studied by Thomas Schwarz, Ph.D., of Children’s Hospital, Boston. These special proteins may be bringing the mitochondria to the terminal where they refine the connection to muscle in a developing nerve. Particular molecules, with the odd names of milton and miro, may very well tell an axonal motor when to unload cargo and build a synapse. The synapse is the specialized, ordered zone for message flow between nerve and muscle.
The synapse could turn out to be where the molecular damage first takes place in mice expressing the SOD1 mutation, although this remains an open question and was a point of lively debate among participants. More on the special process of building a synapse, with guidance by a protein abbreviated as MuSK, was presented by Steven Burden, Ph.D., of New York University. Edwin Levitan, Ph.D., of the University of Pittsburgh, gave details of how packaged proteins arrive at and are captured by the synapse, and Louis Reichardt, Ph.D., of the University of California, San Francisco, had additional insights on the role of special proteins that construct the synapse.
Mobley of Stanford detailed a way to study the flow of materials in axon terminals by growing nerve cells in microscopic channels etched into silicon chips. This is a cutting-edge technique called microfluidics. Each of the cellular packets called endosomes can be tagged by a fluorescent molecule and filmed under a microscope, moving in real time along the axon. Loss of the endosome that carries the helper molecule, nerve growth factor, may be the source of the problem in Down’s syndrome, Mobley’s findings suggest.
Gerardo Morfini, Ph.D., who is working with funding from The ALS Association at the University of Illinois, Chicago, and collaborators showed that mutant SOD1 slows transit of packages in a squid model of axonal transport. The mutant SOD1 proteins inhibit axonal transport by selectively activating a signaling pathway (controlled by proteins called kinases). Morfini and collaborators also are finding specific compounds which can prevent these effects in squid axons, providing the basis for a novel therapeutic approach in ALS.
Gary Banker, Ph.D., of Oregon Health and Science University, Portland, showed that proteins need to be in specific places inside cells and are placed with the aid of marking molecules. Some of these target only axons. This leads to the idea of a smart engine, which recognizes a specific protein in the axon’s transport system. Again, this could open a way to therapeutics in ALS.
|
axonal_trafficking.mov (1.8mb) |
| This neuron expresses a tagged protein (NgCAM) that turns up exclusively on the surface of axons. Carriers containing this axon protein travel freely into all the processes radiating from the cell body, the axon (top right) and all dendrites. Carriers travel out from and back to the cell body. The bright spot in the cell body corresponds to the Golgi area where the carriers are made. --modified from Gary Banker lab home page |
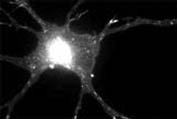
Fuel for Synapses and Protein Synthesis
The mitochondria move along the axon, but scientists did not think they were able to divide and proliferate there. Nor did scientists think that protein assembly could take place along the axon. Both of these bits of dogma are being cast aside. Peter Hollenbeck, Ph.D., at Purdue, showed that the growing axon concentrates mitochondria at its tip. The helper molecule, nerve growth factor, promotes the pooling of mitochondria at the growth cone. Even if an axon is cut from its cell body, it can live in a dish for a time and mitochondria are still replicating within. This speaks to how repairs might some day be addressed for ALS. Are these mitochondria active or quiescent; can they be induced to aid a sick motor neuron with the proper helper molecules?
Michael Sendtner of the University of Wuerzburg, Germany, who has been investigating the inherited motor neuron disorder, spinal muscular atrophy (SMA), showed that ribosomes, the protein making machinery are in fact present in axons. Persistence of both ribosomes and mitochondria in cut motor axons suggests the damage in ALS might indeed be aided if the right manipulation is discovered.
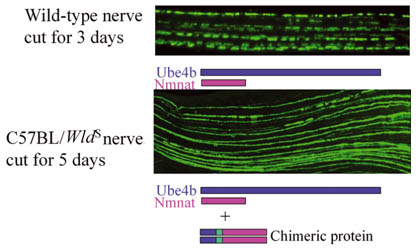
Progress into the role of axon traffic for motor neuron diseases is promised through models of the disease in mice. Two of these, the wobbler mouse, and the mouse with mutation to the gene, Wlds, which protects axons, may provide clues about ALS.
 |
|
Axons from a wild-type, that is, a genetically normal mouse, break into fragments and begin to die after the nerve they are in is cut, but similarly treated axons from a Wlds mutant mouse remain intact for two to three weeks.
Credit: Michael Coleman, Babraham Institute |
Model systems such as these mice, and the squid giant axon preparation of Morfini, provide ways to tease out aspects of the disease process of ALS that could be targeted by therapeutics. A model system to study repair of nerve injury in a living animal as described by Binhai Zheng, Ph.D., of the University of California, San Diego, might give answers for stem cell therapy in ALS. Even certain types of herpes virus which move in restricted fashion along nerves might prove a useful tool to see how axon traffic might be altered in the disease, as presented by Lynn Enquist, Ph.D., of Princeton.
These are just some of the highlights of the workshop. The ALS Association looks forward to renewed efforts and collaborations that will build on the progress in the field of axon dynamics to work toward effective treatment of ALS.