
SOD1 (copper zinc superoxide dismutase 1) and ALS
[Sub topic of Disease Process of ALS]
Related recent news releases
Leads for ALS Therapeutics Inhibit Aggregation of Mutant SOD1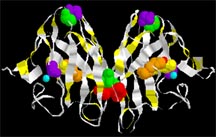 |
| The enzyme, Cu/Zn SOD1, copper-zinc superoxide dismutase. The protein backbone of the molecule is indicated by the white ribbon. The molecules crucial to keeping the molecule in its proper shape are shown as colored balls: these include the copper and zinc ions that help the enzyme do its job of deactivating reactive oxygen. |
Overview Summary
The genetic change in some inherited cases of ALS alters an abundant enzyme within cells, SOD1. A hallmark of ALS is abnormal protein deposits that scientists now know contain mutant SOD1 in the mouse that expresses the human SOD1 gene. But is aggregation a cause, or a bystander effect, of the disease? Further research into the role of SOD1 and of protein aggregates in neurodegenerative diseases should settle the debate and allow for design of appropriate therapies. What is SOD1? In 1993 a large collaborative group demonstrated that some inherited forms of ALS are caused by mutations. The genetic change alters an abundant enzyme within cells called copper-zinc superoxide dismutase (cu-zn superoxide dismutase, now called commonly SOD1). This enzyme serves to keep cells safe from metabolic waste that can do damage if not rendered harmless.SOD1 converts reactive oxygen to harmless water. A copper ion held within the enzyme carries out this chemical change. A zinc atom is also important to the enzyme’s function.
To date, over 100 different mutations in SOD1 have been linked to familial, that is, inherited, ALS. The vast majority of these mutations change the amino acid sequence of the SOD1 protein at a single position. That position is different for most families (see also Genetics of ALS). The most common mutation in North America is the A4V mutation. For an explanation of the genetics of ALS please see information on Genetic Testing.
What is the Role of Mutated SOD1 in ALS? Early studies of mutant SOD1 provided evidence that disease is not caused by loss of this enzyme’s activity. Mice engineered to completely lack SOD1 do not develop ALS. It is now widely accepted that the mutations must cause SOD1 to gain a toxic property. Several reasons for the toxicity are proposed, but none are proven. What are Protein Aggregates? Proteins are the primary ingredient in all biology. The structure of each protein depends on the instructions for its assembly, spelled out in a sequence coded in the genes. First a protein’s amino acids are strung together. Then the chain of amino acids folds up to adopt the proper shape. A mistake in the genetic code, that is, a mutation, places the wrong amino acid in the sequence. This can produce an improperly folded protein that may not work, or that could have improper chemistry that disrupts cellular functions. Or a poorly folded protein can simply be sticky, and gum up the cellular machinery A hallmark of ALS and many other neurodegenerative diseases is abnormal accumulation of proteins called deposits or aggregates. These deposits in mice modeling ALS contain mutant SOD1, researchers now know. Scientists also suspect that mutant SOD1 or some other damaged protein is part of the deposits in ALS.Many different places within the SOD1 protein are altered by the different mutations that researchers have identified in families with inherited ALS. The common aspect to all of these mutations is the disruption of the structure of SOD1. The disrupted SOD1 may become sticky towards itself and towards other proteins. Stopping the aggregation is thus the goal for many researchers in the field.
Trap or Trash? As aggregates form they can trap other proteins that might be maintaining cell health. So it could be that a trap is formed by the mutant SOD1 in ALS, and that is why mutant SOD1 is toxic. Or, the toxicity of aggregates could arise by simply clogging the cellular trash disposal system that handles damaged proteins. Overwhelming the cellular garbage collectors, the proteosomes and the lysosomes, could end up killing the cell.Yet another possibility is that the deposits are simply a feature of dying neurons. Only further research into the role of abnormally accumulated protein in all of the neurodegenerative diseases will settle the debate, and allow for design of appropriate therapies.
