
DISEASE PROCESS OF ALS
Print this page
(requires Adobe Acrobat Reader)
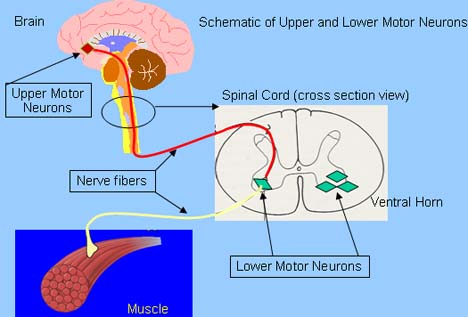
Amyotrophic lateral sclerosis (ALS) kills motor neurons, the large cells of the spinal cord that send nerve fibers out to control the muscles. Also, motor neurons in the part of the brain governing voluntary movements are destroyed in ALS. These so called upper motor neurons send nerve fibers down from the brain to control the lower motor neurons in the spinal cord. Clinicians are also finding that certain cognitive (see section on cognitive changes with ALS) portions of the brain may be affected in ALS as well.
Why are Motor Neurons Affected in ALS?No one knows why the motor neurons are dying in the disease. But researchers have many ideas that lend to speculation. And speculation leads to design of experiments that will find out more about the disease, with funding by The ALS Association (ALSA). Motor neurons must support and supply an incredibly long fiber extended out to make connections. Even though the main part of a motor neuron is of typical size for a cell, its long fiber, called an axon, must travel up to a meter as part of the nerve tracts reaching down to the lower spinal cord, or from the cord out to the fingertips and toes.
Due to the extraordinary metabolic demands of their job, the motor neurons may be particularly vulnerable to any biochemical insult. Motor neurons may be uniquely at the mercy of anything that impedes production of cellular fuel, or impacts cellular supply lines that distribute crucial components to and from the far flung endings of these impressive cells.
Aspects of Motor Neurons Suspect in ALS-
Studies of the proteins within the nerve fibers called axons (see section on axons) that keep them supported and supplied have highlighted their role in maintaining the motor neurons.
-
Prolonged excitation is toxic to nerve cells. Neurobiologists recognize that the nerve cell messenger, glutamate (see section on glutamate), can cause harm when its messages are overwhelming. Abundant evidence points to glutamate as a destructive factor in ALS.
-
There are increasing clues that inflammation contributes to the death of motor neurons in ALS. Evidence suggests that the resident immune cells of the nervous system, the microglia cells, are activated by the disease process of ALS, provoking too much inflammation (see section on inflammation).
-
Mitochondria (see section on mitochondria) are the power plants of all animal cells, and must be especially busy in motor neurons. Indeed, changes in these cell organelles are evident before one can find a physical change such as hind limb weakness in mice that mimic ALS.
-
Programmed cell death is a series of events where a cell essentially shuts itself down. The stepwise self destruction called apoptosis (see section on apoptosis) is an orderly way to remove cells that are damaged beyond repair. In ALS, interrupting apoptosis might prove a way to intervene in the destructive process of the disease.
-
Mutations can produce an improperly folded protein that disrupts cellular functions. Some inherited forms of ALS are caused by mutations in the protein SOD1 (see section on SOD1).
In 1993 a large collaborative group demonstrated that in some inherited cases of ALS a genetic change alters an abundant enzyme within cells called copper-zinc superoxide dismutase 1 (CuZnSOD1, now usually called SOD1). This enzyme serves to keep cells safe from metabolic waste that can do damage if not rendered harmless. A mistake in the genetic code, that is, a mutation, places the wrong amino acid in the sequence for SOD1.
Mice that scientists engineered to express the mutant gene for SOD1 are among the laboratory models (see section on laboratory models of ALS) for research into the causes and avenues towards treatment of ALS.
The fact that these mice recapitulate so many features of ALS gives hope that all cases of ALS can be addressed by the common features that come to light through study of the SOD1 mutation. ALSA funded researchers at the same time are searching relentlessly for other genetic (see section on genetics of ALS) and environmental (see section on environmental factors) factors that might explain the disease and lead clinicians to better therapy.
Related ALSA Funded Research Projects